

CME INDIA Case Presentation by Dr. Kamaldeep Chawla, MD, DNB CARD, FRCP Edinburg, D.M. Sc, Sr. Interventional Cardiologist, Director, Ayushman Heart and Wellness, Vadodara, Dr. N. K. Singh, MD, Diabetes Care Physician, Dhanbad, Dr. Akashkumar N. Singh, MD, Director Spandam Multispeciality Hospital, Vadodara, Vaishnavi Suresh Meagher, MA, Msc, PGDC, Manager Genetic Counselling- Wellytics, Executive Committee Member- Board of Genetic Counselling India, Board Certified Level 2 Genetic Counsellor, Certified Trainer by Health Sector Skill Counsel India, Hyderabad.
CME INDIA Case Study
Abstract
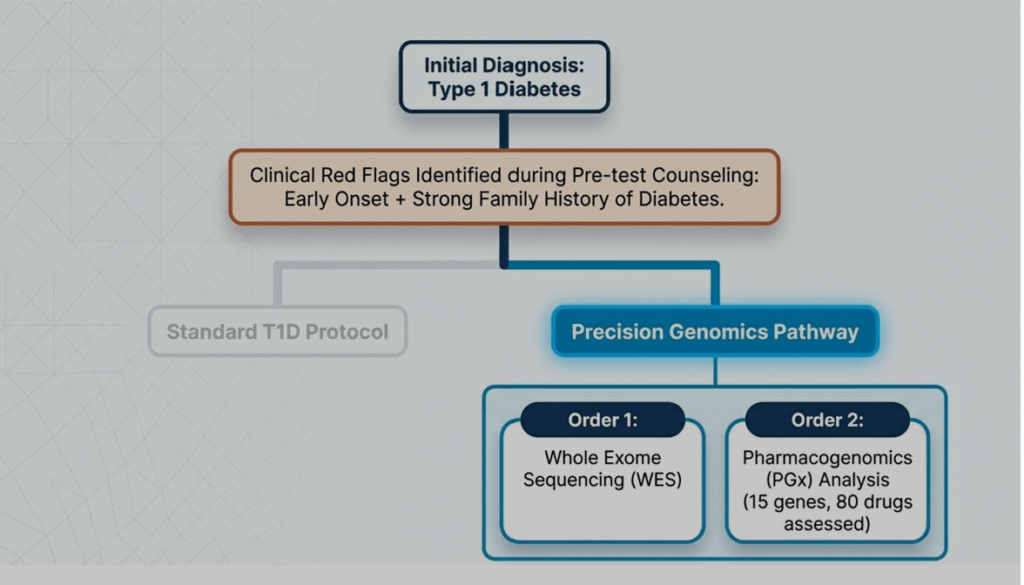
- Monogenic diabetes accounts for 1–5% of pediatric diabetes cases but is frequently misdiagnosed as type 1 diabetes mellitus (T1DM), leading to inappropriate lifelong insulin therapy and delayed recognition of extra-pancreatic manifestations.
- We report an 8-year-9-month-old Indian boy who presented with severe diabetic ketoacidosis (DKA) and was initially diagnosed with T1DM. Prompt referral for whole-exome sequencing (WES) plus pharmacogenomics (PGx) analysis, prompted by early onset and positive family history, identified a novel heterozygous splice-acceptor variant in HNF1B (NM_000458.4: c.1207-1G>A, exon 6), classified as likely pathogenic (ACMG PVS1+PM2). This established the diagnosis of HNF1B-MODY5 / renal cysts and diabetes syndrome (RCAD; OMIM 137920).
- Autoimmune markers were negative, and renal ultrasound showed mild bilateral renal cysts, confirming syndromic correlation. PGx profiling revealed clinically actionable phenotypes: CYP2D6 intermediate metabolizer (IM), TPMT IM, and UGT1A1 IM, prompting immediate medication adjustments and lifelong prescribing alerts. Cascade testing identified the variant in an asymptomatic sibling, enabling pre-symptomatic surveillance. The case underscores the transformative role of comprehensive genomic testing in pediatric diabetes.
- Reclassification from T1DM to MODY5 altered management from insulin-centric protocols to a multidisciplinary MODY5 framework emphasizing renal, hepatic, and uric-acid surveillance. PGx added independent clinical utility by mitigating future drug toxicity risks.
- With a combined diagnostic yield exceeding 30% in selected early-onset cohorts, WES+PGx should be considered early in atypical pediatric diabetes, particularly in consanguinity-low populations with familial clustering. Long-term follow-up will clarify genotype–phenotype correlations for this novel variant.
Introduction
- Maturity-onset diabetes of the young (MODY) comprises a heterogeneous group of autosomal-dominant, non-autoimmune monogenic forms of diabetes typically presenting before age 25 years. Among the 14 recognized subtypes, MODY5 (HNF1B-related) is unique for its multisystem involvement due to the critical role of hepatocyte nuclear factor-1β (HNF1B/TCF2) in embryonic development of the pancreas, kidneys, liver, and genital tract.
- Pathogenic HNF1B variants cause renal cysts and diabetes syndrome (RCAD), characterized by variable renal dysplasia, early-onset diabetes (often insulin-requiring), hyperuricemia, abnormal liver enzymes, and genitourinary malformations. Prevalence estimates vary geographically; in European cohorts, HNF1B mutations explain ~1–2% of MODY, but data from South Asian populations remain sparse. Unlike polygenic T1DM, MODY5 arises from a developmental defect rather than β-cell autoimmunity, rendering autoimmune serology negative and C-peptide preservation possible even after DKA. Misdiagnosis leads to unnecessary insulin dependence, recurrent DKA risk, and failure to screen for extra-pancreatic complications that can progress silently to end-stage renal disease or hepatic fibrosis.
- Recent guidelines from the International Society for Pediatric and Adolescent Diabetes (ISPAD) and the American Diabetes Association recommend genetic testing in children with diabetes onset <12 months or with syndromic features/family history. Whole-exome sequencing (WES) now offers simultaneous interrogation of monogenic diabetes genes and pharmacogenomic (PGx) loci, enabling one-test precision diagnosis and therapy optimization.
- This report details the first documented Indian pediatric case of a novel HNF1B splice variant presenting as severe DKA, reclassified by WES, with integrated PGx data informing lifelong prescribing. It highlights the diagnostic odyssey avoidance, management shift, and cascade-testing benefits achievable through genomic medicine in resource-limited settings.
Case Presentation
An 8-year-9-month-old male child from a non-consanguineous Indian family presented to the emergency department in severe DKA (pH 7.05, bicarbonate 8 mmol/L, blood glucose 512 mg/dL, ketonemia 6.2 mmol/L).
He had a two-week history of polyuria, polydipsia, and 4 kg weight loss. Initial management followed standard pediatric DKA protocol with intravenous fluids, insulin infusion, and electrolyte correction. He stabilized within 48 hours and was discharged on basal-bolus insulin (0.8 U/kg/day).
Autoimmune markers (anti-GAD, anti-IA2) were subsequently negative. Family history was notable for diabetes in both parents (late-onset, non-insulin-dependent) and a maternal grandmother, raising suspicion for monogenic etiology despite the acute DKA presentation.
Pre-test counseling emphasized the possibility of MODY, potential treatment de-escalation from insulin, and implications for family screening.
Written informed consent for WES plus a 15-gene PGx panel (covering ~80 drugs) was obtained.
Whole-exome sequencing
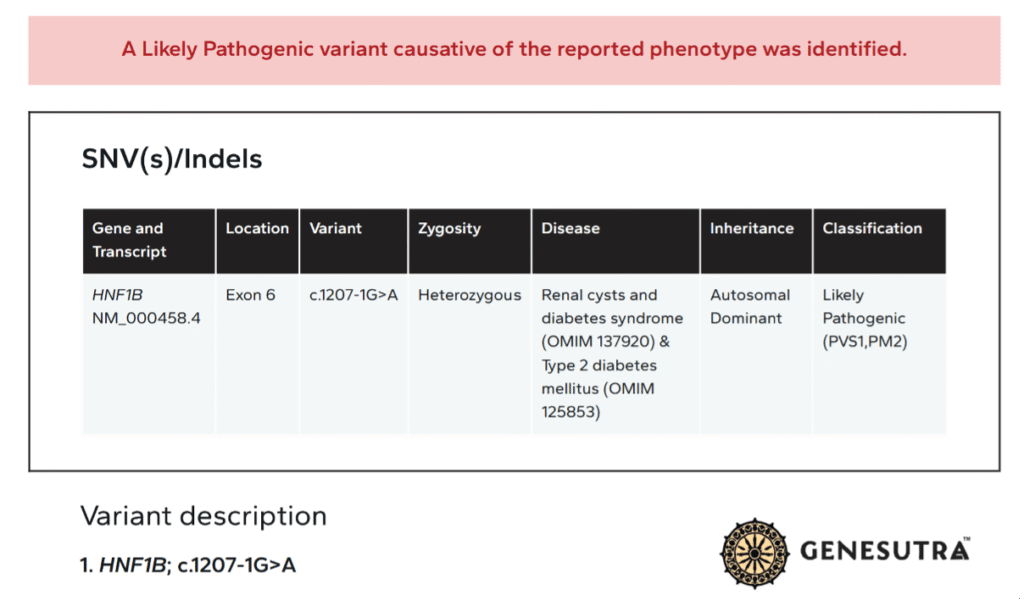
It (Twist 2.0 kit, mean depth 151.76×, 95.98% Q30 bases, 71.08% panel coverage) identified a heterozygous canonical splice-acceptor variant in HNF1B (NM_000458.4: c.1207-1G>A, exon 6).
The variant was absent from gnomAD, 1000 Genomes, and ExAC; in silico tools (MutationTaster, MetaLR, REVEL) predicted deleterious splicing disruption. Per ACMG/AMP guidelines, it met PVS1 (null variant in a gene where loss-of-function is a known disease mechanism) and PM2 (absent from population databases), classifying it as likely pathogenic.
No pathogenic copy-number variants or ACMG secondary/incidental findings were detected.

Supporting clinical correlation:
Renal ultrasound demonstrated mild bilateral renal cysts without structural anomalies; serum uric acid was borderline elevated (6.8 mg/dL); liver enzymes were normal. C-peptide was detectable (0.8 ng/mL), consistent with non-autoimmune etiology.

Concurrent PGx analysis revealed three actionable intermediate-metabolizer phenotypes: CYP2D6 IM → reduced activation of codeine (already prescribed for post-DKA pain) → discontinued and replaced with non-CYP2D6 opioids/NSAIDs.
TPMT IM → increased thiopurine toxicity risk → lifelong alert for 30–50% dose reduction and hematologic monitoring if future immunosuppression required.
UGT1A1 IM → elevated irinotecan toxicity risk → oncology prescribing alert.
Carrier status findings
It included a likely pathogenic HBB splice variant (c.92+5G>C, heterozygous β-thalassemia trait) and a ZNF808 stop-gain (c.2289C>A, heterozygous carrier for pancreatic agenesis-3), plus a known SPINK1 polymorphism (c.101A>G, p.Asn34Ser) conferring 6–12-fold increased risk of chronic pancreatitis in heterozygotes.
These were reported separately with appropriate counselling.
Diagnosis
Post-test counselling revised the diagnosis to HNF1B-MODY5/RCAD. The family was counselled on non-autoimmune inheritance (50% risk to offspring), absence of need for lifelong insulin in some MODY5 cases, and mandatory surveillance. Insulin was continued initially but with a plan for sulfonylurea trial once stable.
Cascade testing of the sibling (age 6 years) confirmed the familial HNF1B variant; the sibling remains euglycemic but entered annual glucose/renal monitoring. Parental segregation studies are pending.
Follow-up
The patient is now under multidisciplinary care (endocrinology, nephrology, genetics) with quarterly HbA1c, annual renal ultrasound/eGFR, uric acid, and liver-function monitoring. PGx results were uploaded as actionable alerts in the electronic health record.
Discussion
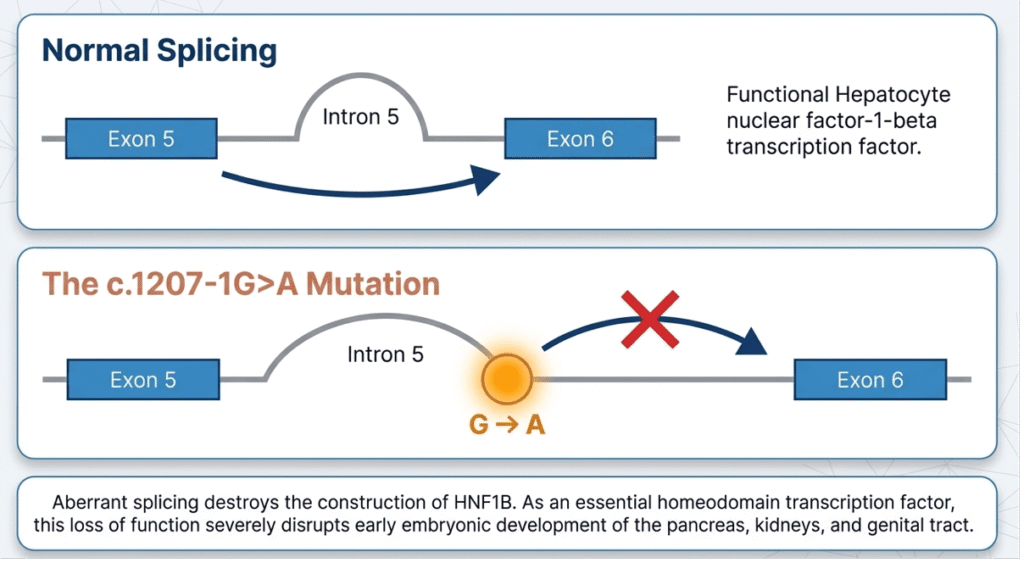
This case exemplifies the diagnostic and therapeutic paradigm shift enabled by next-generation sequencing in paediatric diabetes. The novel HNF1B c.1207-1G>A variant disrupts the canonical splice-acceptor site of exon 6, predicted to cause exon skipping or intron retention, resulting in a non-functional or truncated protein.
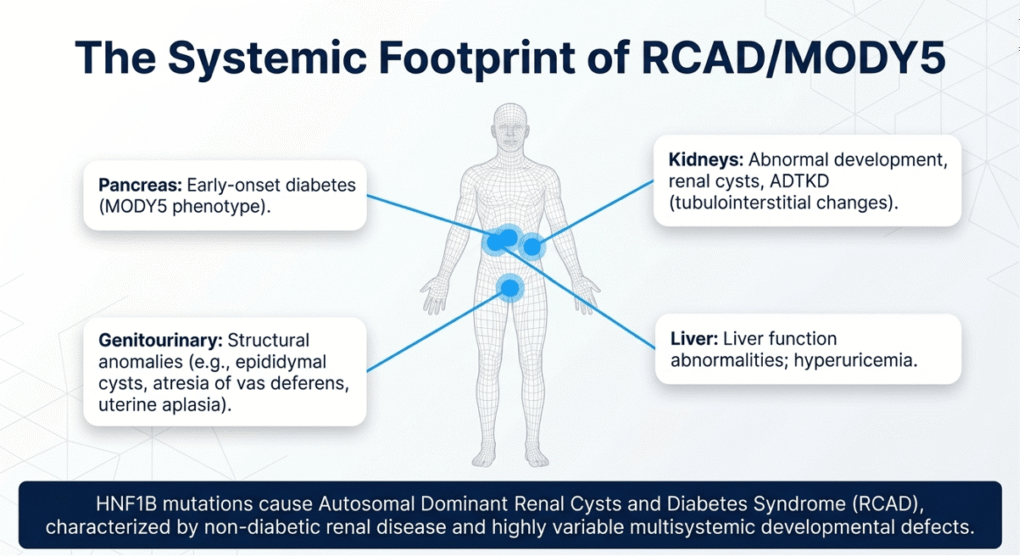
HNF1B is a homeodomain transcription factor expressed from early embryogenesis in the Wolffian duct, metanephric mesenchyme, pancreatic buds, and hepatic diverticulum. Heterozygous loss-of-function therefore produces the pleiotropic RCAD phenotype.
Literature review reveals >200 HNF1B pathogenic variants, predominantly missense or truncating, with marked phenotypic heterogeneity even within families.
Renal involvement occurs in >80% of cases (cysts, hypoplasia, hyperuricemic nephropathy), while diabetes penetrance reaches 50–60% by age 25. Extra-pancreatic features (genital malformations, hypomagnesemia, abnormal LFTs) are present in 30–50%. The present patient’s mild renal cysts and borderline hyperuricemia align with the milder end of the spectrum, yet the severe DKA presentation at age 8.9 years is noteworthy.
Prior reports document MODY5 diabetes onset as early as 10 years (Horikawa et al., 1997); this case extends the lower age limit and demonstrates that DKA, traditionally considered T1DM-specific, can be the sentinel event in MODY5 when β-cell mass is critically reduced. Negative GAD/IA2 antibodies and preserved C-peptide further distinguish it from autoimmune T1DM.
WES proved superior to targeted MODY panels by simultaneously capturing the HNF1B variant and PGx loci. The 15-gene PGx panel identified three IM phenotypes with immediate and future clinical impact. CYP2D6 IM status prompted codeine discontinuation, averting potential opioid inefficacy or toxicity.
TPMT and UGT1A1 alerts will guide future therapy in this child who may require immunosuppression (e.g., post-renal transplant) or oncology care, given the elevated lifetime cancer risk reported in some HNF1B carriers. Integration of PGx into the electronic record fulfills CPIC guideline recommendations and exemplifies “one-test” precision medicine.
Secondary carrier findings merit brief discussion. The HBB variant confirms β-thalassemia trait (prevalent in South Asia); hematologic monitoring is routine. Heterozygous ZNF808 loss-of-function is unlikely to cause phenotype given recessive inheritance of pancreatic agenesis-3. The SPINK1 p.Asn34Ser polymorphism, enriched in Indian populations, confers modest pancreatitis risk but requires a “second hit” (genetic or environmental); alcohol avoidance and triglyceride control were counseled. These incidental findings illustrate the added value—and counseling burden—of WES over targeted panels.
Management was transformed from a T1DM insulin-only paradigm to a MODY5 framework: (1) continued insulin with future sulfonylurea or GLP-1 receptor agonist trial once glycemic stability is achieved (HNF1B patients retain some β-cell responsiveness); (2) lifelong nephrology surveillance to prevent progression of cystic disease to chronic kidney disease; (3) annual uric acid and liver-function tests; (4) family cascade testing. Early sibling diagnosis enables pre-symptomatic intervention, potentially delaying diabetes onset through lifestyle measures.
Limitations of WES include incomplete coverage of deep intronic/regulatory regions and inability to detect large rearrangements or mitochondrial variants; however, the high-quality metrics (>95% at 20×) and orthogonal clinical correlation mitigate these concerns. Variant reclassification remains possible with functional studies (minigene splicing assays) or additional family segregation.
Broader implications: In India, where T1DM incidence is rising and monogenic diabetes is under-diagnosed, cost-effective WES (now comparable to lifelong insulin complications) should be prioritized in selected cases—early onset, family history, negative autoantibodies, or syndromic clues. Health-economic modeling suggests that genomic reclassification reduces lifetime costs by 20–40% through de-intensified insulin regimens and complication prevention. National guidelines should incorporate reflex WES+PGx for atypical pediatric diabetes.
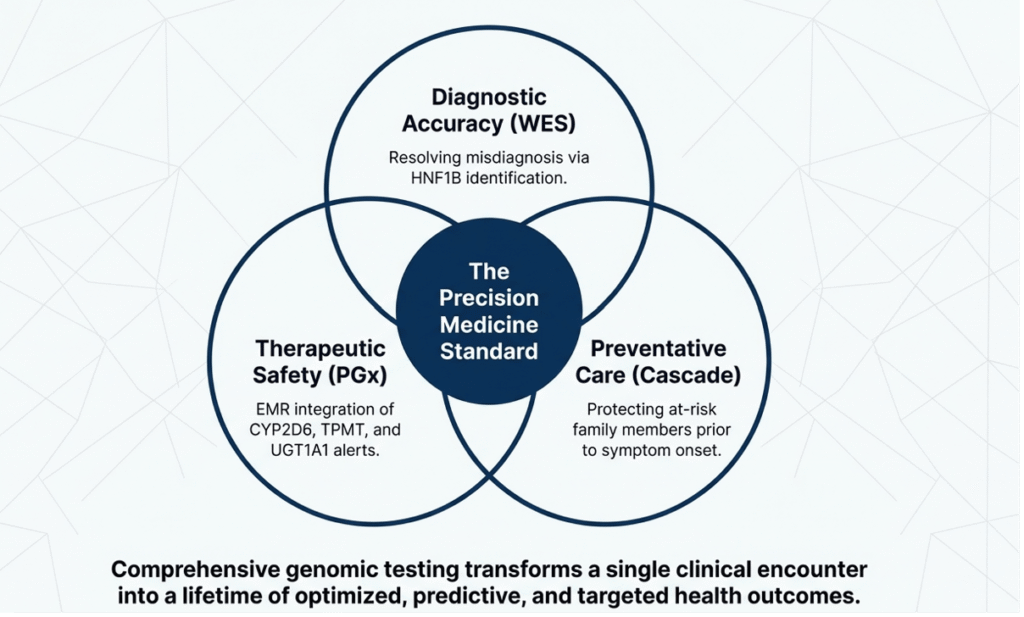
Conclusion:
This case illustrates the power of WES to reclassify severe DKA in childhood from presumed T1DM to HNF1B-MODY5 caused by a novel splice variant. Integration of PGx data provided independent therapeutic guidance, while cascade testing enabled pre-symptomatic family care. Comprehensive genomic testing in early-onset familial diabetes facilitates accurate diagnosis, precision therapy, surveillance of extra-pancreatic manifestations, and cost-effective long-term management. As genomic literacy grows and sequencing costs decline, such “one-test” strategies will become standard, ultimately reducing the global burden of misdiagnosed monogenic diabetes
References:
- Horikawa Y, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384-5.
- Edghill EL, et al. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. 2006;43(1):84-90.
- Fajans SS, et al. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971-80.
- De Franco E, et al. Primate-specific ZNF808 is essential for pancreatic development in humans. Nat Genet. 2023;55(12):2075-2081.
Acknowledgements: The authors thank the family for consenting to publication and the referring clinician for multidisciplinary coordination. No funding was received for this report. Conflicts of Interest: None declared. Ethics: Written informed consent obtained; report prepared in accordance with CARE guidelines for case reports. All figures were created on Notebook LM. AI has been used for language modifications only.
Discover CME INDIA:

- Explore CME INDIA Repository
- Examine CME INDIA Case Study
- Read History Today in Medicine
- Register for Future CMEs