

CME INDIA Case Presentation by Prof. Dr. G. Prakash, MD (Gen. Med), D. Diab, Professor and Head, Dept. of Diabetology, Govt. Mohan Kumaramangalam Medical College, Salem Tamil Nadu.
CME INDIA Case Study
How Presented?
- A 15-year-old female was referred from General surgical OPD with complaints of abdominal pain on and off for past 1 year with uncontrolled blood sugar levels.
- She was diagnosed as a case of Diabetes mellitus before 1 year on Oral Hypoglycemic agents (Metformin+ Glimepiride 1mg) (on irregular treatment).
- No H/o similar illness in family members (born out of non-consanguineous marriage).
- Attained menarche at 13 years of age.
- Irregular cycles, no menstrual flow for past 1 year.
What examination showed?
- Patient was conscious, oriented emaciated
- No pallor
- Not icteric
- No cyanosis/clubbing
- No generalized lymphadenopathy
- No pedal edema
- CVS-S1S2+
- RS-B/L AE+
- P/A- Distended, not tender
- Anthropometry
- Height – 144cm
- Weight- 35 kg
- BMI- 16.9
- Waist circumference – 74
- Hip circumference – 70
- WHR – 1.057
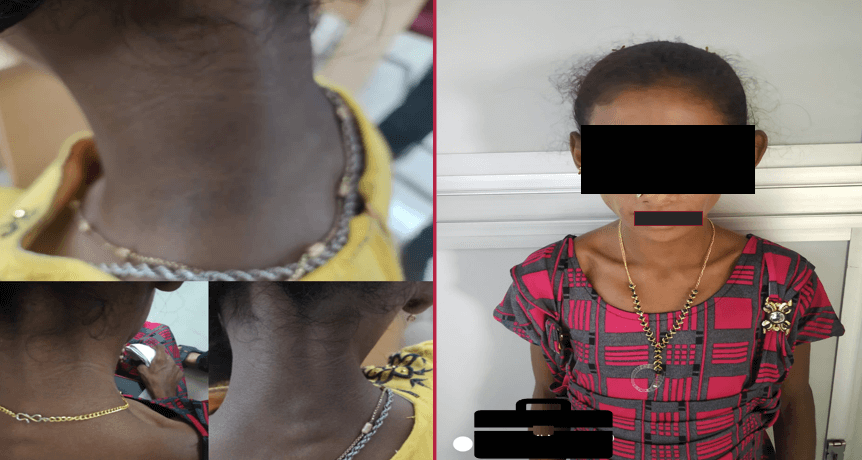
Findings:
- Loss of buccal pad of fat.
- Acanthosis nigricans (nape of neck, B/L axilla).
- Eruptive xanthoma (in dorsum of hands and dorsum of elbow joint).
- Muscular appearance (no fat).
- Abdominal distension with mild umbilical hernia.
- Loss of secondary sexual characters.
Investigations:
| TC | 12100 |
| Hb | 14.1g/dl |
| Platelets | 1.98 lks |
| RBS | 390 mg% |
| Acetone | Negative |
| HbA1c | 16.1% |
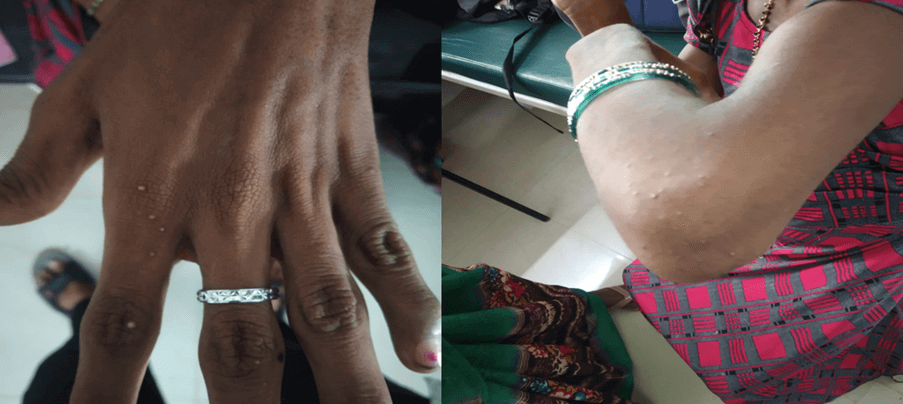
With the above investigations, patient was started on Insulin (20 units total)
- She was asked to come for follow up.
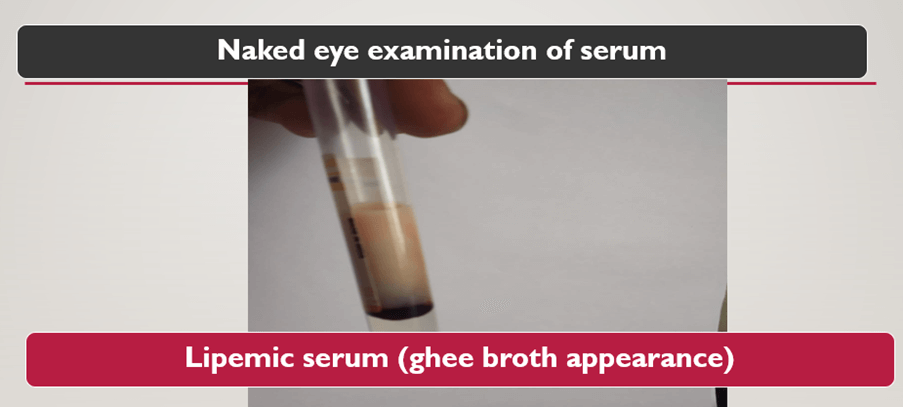
| Total cholesterol – 580mg% |
| Triglycerides – >2000 mg% |
| HDL- 30 mg% |
| LDL – 360 mg% |
| Urea – 16 |
| S.Cr – 0.6 |
| Urine Routine- albumin(nil) |
| Amylase -26 |
| Lipase – 12 |
| Fasting Insulin – 47.25 µU/ml (0-25) |
| FBS – 401 |
| HOMA IR – 46.8 (0.7-2) |
| Fasting C peptide-2.57 ng/ml (0.5-2.7) |
| TSH-3.4 |
| FT4-1.1 |
USG of abdomen and pelvis
- Liver – Hepatomegaly- 15.2 cm.
- Grade 2 fatty liver.
- Spleen (normal size and echotexture).
- Kidneys normal.
- Normal pancreas.
- Ovaries – No PCOS.
- No free fluid.
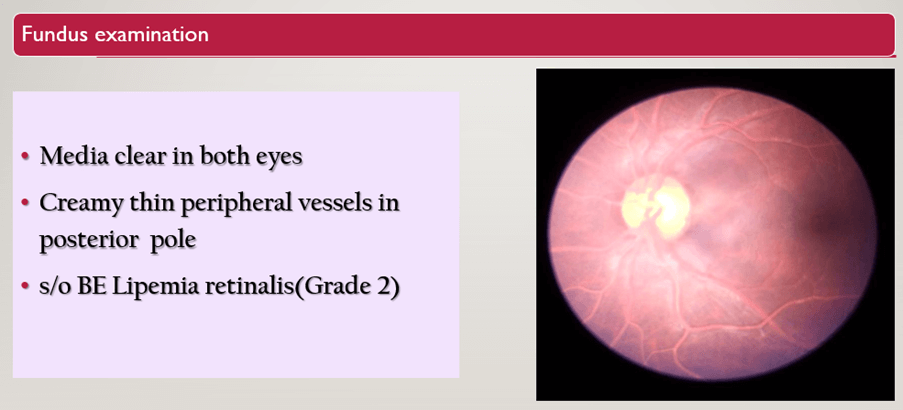
Fundus Examination
- It shows Media clear in both eyes.
- Creamy thin peripheral vessels in posterior pole.
- Suggestive of BE Lipemia retinalis(Grade 2).
What is this case?
- 15 yr. old female diagnosed diabetes mellitus presented with h/o recurrent abdomen pain, no h/o steatorrhea, no h/o previous admissions for DKA, no family h/o Diabetes.
- Malnourished with muscular prominence, acanthosis nigricans, with features of hypertriglyceridemia and low HDL cholesterol.
How Managed?
- Initially She was started on Conventional Insulin 20 units /days then gradually increased to 105 units/day(3U/kg/day).
- In spite of high doses of insulin, her blood sugar remained uncontrolled, so she is started on Pioglitazone and blood sugar still uncontrolled. She had metformin intolerance. Saroglitazar was also started.
Diagnosis
| Diabetes mellitus (Age at onset – adolescence) |
| Severe Insulin resistance |
| Eruptive xanthomas |
| Hypertriglyceridemia |
| Lipemia retinalis |
| Lipodystrophy |
| These features constitute Generalized lipodystrophy diabetes mellitus |
Could it be Berardinilli – Seip syndrome?
- Autosomal recessive disorder.
- Complete absence of subcutaneous fat from birth, (muscular appearance).
- Lipids are stored in metabolically active tissues.
- Those affected have features of severe insulin resistance including often widespread acanthosis, hypertriglyceridemia, and low HDL cholesterol.
- Hepatic steatosis occurs early and may lead to cirrhosis; hepatomegaly is seen frequently.
- Childhood growth is accelerated and bone age advanced. Diabetes commonly develops during adolescence.
- Other associated features include acromegaloid features, hypertrophic cardiomyopathy, skeletal muscle hypertrophy, bone cysts, and intellectual impairment.
- Serum leptin and adiponectin levels are markedly reduced.
What is Congenital generalized lipodystrophic diabetes mellitus?
- Three molecularly distinct forms have been identified:
- Congenital generalized lipodystrophy types 1, 2 and 3 resulting from mutations in 1-acyglycerol 3-phosphate-O-acyltransferase 2 (AGPAT2), Berardinelli-Seip congenital lipodystrophy 2 (BSCL2), and Caveolin-1 (CAV1).
- Some people with this phenotype do not have mutations in any of these genes.
- Other inherited forms of lipodystrophy rare subtypes of lipodystrophy associated with dysmorphic features include mandibuloacral dysplasia (lipodystrophy with characteristic skeletal abnormalities), SHORT syndrome (short stature, hyperextensibility of joints, ocular depression, Reiger anomaly, teething delay) and neonatal progeroid syndrome.
Diagnostic criteria
- Berardinelli-Seip congenital lipodystrophy (BSCL) should be suspected in individuals with one or more of the following major and/or minor findings.
Major Criteria
- Lipoatrophy affecting the trunk, limbs, and face- athletic appearance, skeletal muscle hypertrophy is also present.
- Acromegaloid features include gigantism, muscular hypertrophy, advanced bone age, prognathism, prominent orbital ridges, enlarged hands and feet, clitoromegaly, and enlarged external genitalia in males.
- Hepatomegaly.
- Elevated serum concentration of triglycerides.
- Insulin resistance. Elevated serum concentrations of insulin and C-peptide may occur starting in the first years of life. Its early clinical expression is acanthosis nigricans of the groin, neck, and axillae.
Minor Criteria
- Hypertrophic cardiomyopathy.
- PsychoMinormotor retardation or mild (IQ 50-70) to moderate (IQ 35-50) intellectual impairment.
- Hirsutism.
- Precocious puberty in females.
- Bone cysts observed in individuals with biallelic pathogenic variants in AGPAT2.
- Phlebomegaly.
Management Pearls
- Lifestyle changes (extremely low-fat diet <15% total energy from fat, increased physical activity).
- Fibrates.
- Glycemic control (Metformin/Pioglitazone+ High dose insulin therapy).
- Leptin replacement (Metreleptin).
Approach Consideration
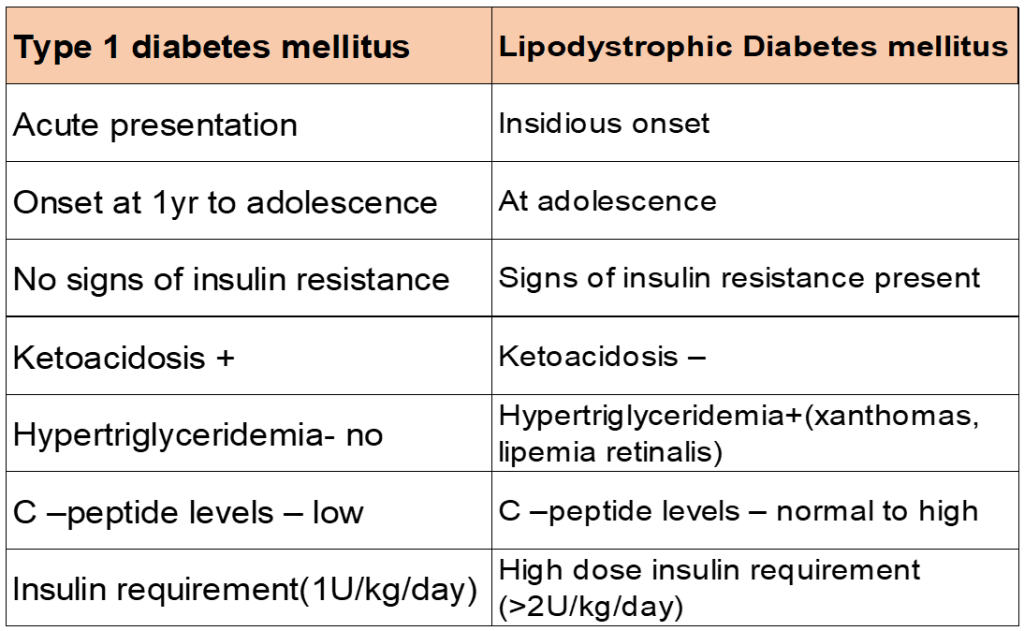
Differentiating T1DM with Lipodystrophic DM
CME INDIA Learning Points
- Based on the overall presentation, the diagnosis of Generalized Lipodystrophic Diabetes Mellitus (GLDM) seems likely.
- GLDM is a rare genetic disorder characterized by a deficiency of adipose tissue, leading to severe insulin resistance, hypertriglyceridemia, eruptive xanthomas, and other metabolic abnormalities like lipemia retinalis. The onset of symptoms typically occurs during adolescence. The presence of lipodystrophy further supports this diagnosis.
- The clinical presentation resembles that of Berardinelli-Seip syndrome, although lipoatrophy typically emerges later in childhood, adolescence, or adulthood, suggesting an acquired nature for the syndrome. In some instances, adipose tissue loss is localized, particularly if preceded by panniculitis. Patients often exhibit an increased appetite and accelerated growth during adolescence. About a third of cases may display acanthosis nigricans and polycystic ovary syndrome. Hepatomegaly with steatosis and a risk of cirrhosis progression are common findings. Biochemically, hyperinsulinemia and insulin-resistant diabetes are prevalent, often accompanied by severe hypertriglyceridemia and decreased plasma levels of leptin and adiponectin.
- Recent reports indicate proteinuria linked with focal segmental glomerulosclerosis or membranoproliferative glomerulonephritis, along with growth hormone dysregulation. The syndrome manifests in three types: 1) featuring panniculitis (inflammatory nodules followed by lipoatrophy), 2) an autoimmune variant often associated with syndromes such as chronic active hepatitis, Hashimoto’s thyroiditis, and hemolytic anemia, as well as dermatomyositis and Sjogren’s syndrome, and 3) idiopathic.
- The exact cause of the disease remains elusive. It’s suggested that infectious triggers, such as in a recent case where the panniculitis type followed tuberculosis, or autoimmune mechanisms may play a role. A recent study demonstrated activation of the classical complement pathway, as evidenced by low levels of C4. This differs from acquired partial lipodystrophy, which primarily affects the upper body and is associated with activation of the alternative complement pathway (indicated by low C3 levels). Progression to partial lipoatrophy, either focal or generalized, has been observed in patients with dermatomyositis, particularly in cases with late relapses, often coinciding with the presence of the anti-p155 antibody. The possibility of an underlying genetic component has not been ruled out.
- The diagnosis primarily relies on clinical evaluation and should be confirmed through assessments of body fat, notably via biphotonic absorptiometry and magnetic resonance imaging.
- Determining whether a patient has lipodystrophy involves recognizing its distinct features. Generalized Lipodystrophy (GLD) typically presents with conspicuous characteristics, while Partial Lipodystrophy (PLD) may exhibit more subtle signs, often characterized by a specific pattern of fat loss. Lipodystrophy can manifest in childhood or adulthood, with onset being sudden or gradual. Importantly, fat loss in lipodystrophy is irreversible, except in rare instances.
- Lipodystrophy should be suspected when patients display congenital or progressive deficiency of subcutaneous adipose tissue (SAT), often accompanied by autoimmune diseases, fat loss in limbs combined with fat accumulation elsewhere, or SAT deficiency alongside other physical abnormalities. Additional features may include failure to thrive (in children), prominent muscles and veins, acanthosis nigricans, eruptive xanthomas, or a cushingoid or acromegaloid appearance. The presence of diabetes mellitus requiring high insulin doses, severe hypertriglyceridemia, nonalcoholic steatohepatitis, or polycystic ovarian syndrome (PCOS) further suggests lipodystrophy.
- Diagnosing lipodystrophy relies on clinical history, physical examination, and body composition assessment, with laboratory tests providing additional insights in some cases. While no definitive diagnostic criteria based on skinfold measurements or imaging procedures exist, evaluations such as dual-energy X-ray absorptiometry and magnetic resonance imaging aid diagnosis. Although serum leptin levels in lipodystrophy patients are typically low, no specific threshold can definitively rule out the diagnosis.
- Differential diagnoses encompass other conditions characterized by severe insulin resistance, such as Rabson-Mendenhall syndrome, leprechaunism, Berardinelli type lipodystrophy, and insulin resistance syndromes types A and B.
- Additionally, other forms of lipodystrophies should be considered during the diagnostic process.
- The management of the metabolic aspects typically aligns with approaches used for other forms of insulin resistance. This includes lifestyle interventions such as physical exercise, along with medications like metformin or pioglitazone to enhance insulin sensitivity. Insulin or insulin analogues are often prescribed, alongside antihypertensive agents, and interventions for monitoring and managing hypertriglyceridemia. While recombinant human leptin has shown efficacy in improving metabolic parameters, its availability varies across countries. In severe autoimmune variants, immunosuppressive therapy might be warranted.
- Currently, there is no curative treatment available for lipodystrophy syndromes. The primary goal of therapeutic approaches is to alleviate metabolic abnormalities and associated conditions resulting from lipodystrophy. Plastic surgical interventions may be considered if necessary.
- Managing metabolic disturbances associated with lipodystrophy, such as insulin resistance, diabetes, hypertriglyceridemia, and fatty liver disease, typically involves dietary and exercise interventions, along with pharmacotherapy. This may include medications like antidiabetic and antidyslipidemia agents (e.g., Metreleptin/Leptin replacement therapy, insulin secretagogues, fibrates, statins, eicosapentaenoic acid, insulin-like growth factor 1).
- While there is currently no cure for adipose tissue atrophy, thiazolidinedione medications can induce hypertrophy of adipose tissue in non-atrophic areas, but do not affect the areas already affected by atrophy. However, in cases of acquired lipodystrophy syndrome, strategies targeting the underlying condition, such as subcutaneous panniculitis or autoimmune disease, may be effective.
- Plastic surgery options include facioplasty involving autologous transplantation of subcutaneous fat tissue, free flap procedures, or implantation of silicone or other substitutes for fat tissue. Soft-tissue expansion using adipose tissue-derived stem cells is still being researched, and its safety and efficacy are yet to be determined.
- Regarding prognosis, it’s not extensively documented but is likely influenced by cardiovascular risk factors associated with insulin resistance and the underlying etiology of the disease. Regular monitoring and comprehensive management are essential for optimizing outcomes.
CME INDIA Quick Take-Away
- Congenital generalized lipodystrophy (Berardinelli-Seip syndrome) is an autosomal recessive disorder marked by a nearly complete absence of fat from birth or infancy, alongside notable muscle prominence, phlebomegaly, acanthosis nigricans, hepatomegaly, umbilical prominence, and a strong appetite during childhood. Various genetic causes have been identified, each presenting distinct clinical features. Metabolic complications are common and can be severe, sometimes leading to cardiomyopathy or rhythm disturbances.
- Familial partial lipodystrophy (FPLD) encompasses a group of usually autosomal dominant disorders characterized by fat loss in specific regions such as limbs, buttocks, and hips. Regional fat accumulation, varying by subtype, may give rise to a Cushingoid appearance. Metabolic complications often emerge in adulthood, increasing the risk of coronary heart disease and occasionally early cardiomyopathy.
- Acquired generalized lipodystrophy (Lawrence syndrome), more prevalent in females, typically manifests before adolescence with progressive fat loss throughout the body, including palms and soles. Some fat accumulation may occur in the face, neck, or axillae. Metabolic complications are frequent and can be severe, often co-occurring with autoimmune diseases.
- Acquired partial lipodystrophy (Barraquer-Simons syndrome), more common in females, usually begins in childhood or adolescence, characterized by fat loss following a cranio-caudal pattern. Fat accumulation may appear in the hips, buttocks, and legs. It is often linked with autoimmune diseases, particularly membranoproliferative glomerulonephritis. Metabolic complications are less common.
- The diagnosis of lipodystrophy relies on thorough history-taking, physical examination, assessment of body composition, and metabolic status. While there are no specific serum leptin levels defining or excluding lipodystrophy, confirmatory genetic testing is beneficial in suspected familial cases, and it should be considered for at-risk family members. Serum complement levels and autoantibodies may support the diagnosis of acquired lipodystrophy syndromes. However, definitive diagnostic criteria for lipodystrophy have yet to be established.
CME INDIA Tail Piece
Metreleptin:
- In generalized lipodystrophy, Metreleptin, in conjunction with diet, is recommended as a first-line treatment for managing metabolic and endocrine abnormalities. It may also be considered for preventing these comorbidities in children.
- For individuals with partial lipodystrophy who have low leptin levels (leptin <4 ng/mL) and severe metabolic disturbances (HbA1c >8% and/or triglycerides >500 mg/dL), Metreleptin may be considered.
- Metreleptin, a recombinant human methionyl leptin, is currently the only drug specifically approved for treating lipodystrophy. It is approved in the United States for managing metabolic complications in patients with generalized lipodystrophy and in Japan for both generalized and partial lipodystrophy. Compassionate use programs make it accessible in other regions like Europe. There is no age restriction for initiating Metreleptin therapy; even infants as young as 6 months have received treatment. Dosing adjustments should be tailored according to metabolic parameters and changes in weight, with clinical and laboratory assessments recommended every 3 to 6 months.
- The cost of Metreleptin can vary widely depending on factors such as the country of purchase, healthcare coverage, dosage, and treatment duration. In the United States, where it is approved for the treatment of metabolic complications in patients with generalized lipodystrophy, Metreleptin can be quite expensive, with an annual cost ranging from tens of thousands to over a hundred thousand dollars. However, it’s essential to note that many factors influence the actual cost an individual may incur, including insurance coverage, discounts, patient assistance programs, and negotiated prices with healthcare providers or pharmacies.
- In other countries where Metreleptin may be available through compassionate use programs or under different regulatory approvals, the cost structure may vary. It’s advisable for individuals seeking information on the cost of Metreleptin to consult with their healthcare provider, insurance company, or local pharmacy for specific pricing details and potential financial assistance options.
References:
- Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, Mungai L, Oral EA, Patni N, Rother KI, von Schnurbein J, Sorkina E, Stanley T, Vigouroux C, Wabitsch M, Williams R, Yorifuji T. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016 Dec;101(12):4500-4511. doi: 10.1210/jc.2016-2466. Epub 2016 Oct 6. PMID: 27710244; PMCID: PMC5155679.
- Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step-by-step approach. J Endocrinol Invest. 2019 Jan;42(1):61-73. doi: 10.1007/s40618-018-0887-z. Epub 2018 Apr 27. PMID: 29704234; PMCID: PMC6304182.
- Mondal N, Mandal S, Nandy P, Naskar B. Barraquer-Simons syndrome: an unusual form of acquired partial lipodystrophy in a child with lupus nephritis. Indian Dermatol Online J. 2023 Nov-Dec. 14(6):890-2.
- [Guideline] Tanaka T, Kusakabe T, Ebihara K, Aizawa-Abe M, Aotani D, Yorifuji T, et al. Practice guideline for lipodystrophy syndromes-clinically important diseases of the Japan Endocrine Society (JES). Endocr J. 2021 Sep 28. 68(9):1027-42.
Discover CME INDIA:

- Explore CME INDIA Repository
- Examine CME INDIA Case Study
- Read History Today in Medicine
- Register for Future CMEs
Good case