

CME INDIA Presentation by Dr. N. K. Singh, MD, FICP, FACP, FRSSDI, Diabetologist Physician, Director – Diabetes-Heart Research Centre, Dhanbad, Jharkhand, National EC Member, RSSDI, Editor: CME INDIA.

The Challenge
- We often think only of Type 1 and Type 2 Diabetes.
- Together, they account for 90% of cases.
- However, 10% fall under the category of “Atypical Diabetes.”
- Every practitioner is likely to encounter it. The challenge lies in recognizing these complex cases.
What is Atypical Diabetes?
- It is increasingly acknowledged that the traditional binary classification of diabetes into type 1 and type 2 falls short in encompassing the diverse metabolic and clinical phenotypes, molecular mechanisms, and disease pathways that culminate in hyperglycemia.
- Atypical diabetes is suspected in individuals who do not neatly fit the currently accepted criteria defining type 1 diabetes (T1D), type 2 diabetes (T2D), or secondary diabetes. These cases exhibit unique features that challenge traditional classifications.
- The prevalence of non–type 1 and non–type 2 diabetes—now termed “atypical diabetes”—is probably underestimated, with proportions varying significantly (ranging from 5% to 11%) based on ethnic and other population-specific factors
Snap Shot of a Case
- A 12-year-old-female presented with a history of decreased activity and increased sleeping since 4 months of age. She had no polyuria, polydipsia, or weight loss. At that time, her blood sugar was 535 mg/dL. She was diagnosed with Type 1 diabetes and put on insulin at age 4.
- Now, at age 11, a detailed past history was taken. She had developmental delay, scoliosis, lack of communication and smile, difficulty with latching during feeding, poor muscle tone, inability to hold her head up, and inability to grab objects by age 4. She also had a turned foot. By age 5, she was not walking or talking but could hop and scoot.
- Genetic testing was advised, revealing a KCNJ11 mutation, specifically a heterozygous p.Val 59Ala mutation. She was subsequently shifted to Glibenclamide at a dose of 10 mg TID (1.38 mg/kg). Her HbA1c later came down to 5.5%.
- She has a history of multiple seizures, for which she is on Carbamazepine. Remarkable developmental progress has been observed. This improvement may be due to the role of sulfonylureas (SU) in the KATP channels in the brain. Clinical Pearl:
- Activating mutations in KCNJ11 or ABCC8 generally decrease the channel’s responsiveness to ATP, thereby preventing channel closure and subsequent insulin secretion. The specific mutation dictates the phenotype, and for Kir6.2 mutations, there is a notable correlation between the mutation’s functional severity and the clinical presentation.
- Final Diagnosis: Monogenic diabetes with KCNJ11 V59A mutation, presenting as DEND syndrome (Developmental delay, Epilepsy, Neonatal Diabetes).

History may give vital clues
- A 26-year-old man, at his initial visit, explains that he has had diabetes since he was 18 years old. His father and his only brother were also diagnosed with diabetes in their late teens. All were initially believed to have type 1 diabetes and were started on insulin as initial therapy at the time of diagnosis. The diagnosis seemed certain given their young age, lean build, and white ethnicity, with no signs of insulin resistance, which would have suggested type 2 diabetes.
- This patient’s insulin dose has always been unusually low (less than 10 units per day, with no basal insulin needed). His fasting blood glucose is usually below 110 mg/dL, and he can occasionally miss insulin doses, especially if he eats low-carbohydrate meals. He is prone to hypoglycemia when he increases his insulin or when he exercises.
- No episodes of diabetic ketoacidosis have been reported.
- The patient has a BMI of 23, is normotensive, and shows no signs of acanthosis nigricans, diabetic retinopathy, or neuropathy. His HbA1c is 6.4%. His father has recently stopped all insulin and is now doing well on low-dose glimepiride therapy (1 mg/day). The patient asks, “Do I actually have type 2 diabetes?”
- Given his lean build, and family history, this patient has all the features consistent with a diagnosis of Hepatocyte Nuclear Factor 1α (HNF1A) monogenic diabetes.
- Key characteristics of this type of diabetes include minimal requirement for insulin, optimal fasting glycemic control, and often a favorable response to low-dose sulfonylurea therapy. Individuals with HNF1A diabetes typically have a heightened sensitivity to sulfonylureas, making them an effective treatment option.
- It’s important to monitor for any signs of insulin resistance or episodes of diabetic ketoacidosis, as these can still occur in some cases. Regular monitoring of blood glucose levels and close follow-up with a healthcare provider are essential for managing this condition effectively.
When encountering a patient with a strong multi-generational history of early-onset diabetes and minimal insulin requirements, consider the possibility of an unusual form of diabetes with a strong genetic component, such as MODY type 3 (HNF1A diabetes).
Key Takeaways:
- Genetic Testing: Commercially available genetic testing for common HNF1A mutations can confirm the diagnosis, guiding tailored treatment approaches.
- Treatment Approach: Patients with HNF1A diabetes often exhibit heightened sensitivity to sulfonylureas. Transitioning from insulin to low-dose oral sulfonylurea therapy can be an effective management strategy.
- Long-term Complications: Despite differences in etiology, patients with HNF1A diabetes are at risk for similar long-term complications as those with type 1 diabetes. Regular monitoring and preventive measures are crucial.
- Family Planning: Counseling regarding the genetic nature of the condition is essential for informed family planning decisions, highlighting the importance of genetic testing and implications for future generations.
See this case
- A 12-year-old male was diagnosed with diabetes at the age of 9 following presentation with urinary frequency. His fasting blood glucose ranged from 120–140 mg/dL, and an oral glucose tolerance test revealed a fasting blood glucose of 130 mg/dL and a 2-hour blood glucose of 216 mg/dL. Initially, he was treated with metformin 500 mg once daily, gradually increased to 500 mg twice daily, maintaining stable HbA1c levels around 6.4%.
- His past medical history indicated obesity, with a BMI consistently above the 90th percentile for his age, while pubertal development progressed normally. Family history was notable for multiple maternal relatives with diabetes; his mother had a BMI of 29 and a history of mildly elevated HbA1c, managed with glimepiride, with recent levels at 6.6%. Similarly, his maternal grandfather managed mild hyperglycemia with pioglitazone. His maternal great-grandmother, aged 90, had a history of mild diabetes but was off medication due to hypoglycemia side effects, without complications.
- Social history was unremarkable, and his current medications included metformin 500 mg twice daily, with no reported allergies. Upon examination, his vital signs were stable, with a blood pressure of 109/67 mmHg and a pulse of 75 bpm. His BMI was 28.37 kg/m², with physical signs indicating generalized obesity, Tanner stage 4–5 pubic hair, and pubertal testes. Laboratory results revealed an HbA1c of 6.5%.
- The patient underwent genetic testing, which identified a heterozygous mutation in the GCK gene: c.667G>A (p.Gly223Ser), as revealed by research-based Sanger sequencing

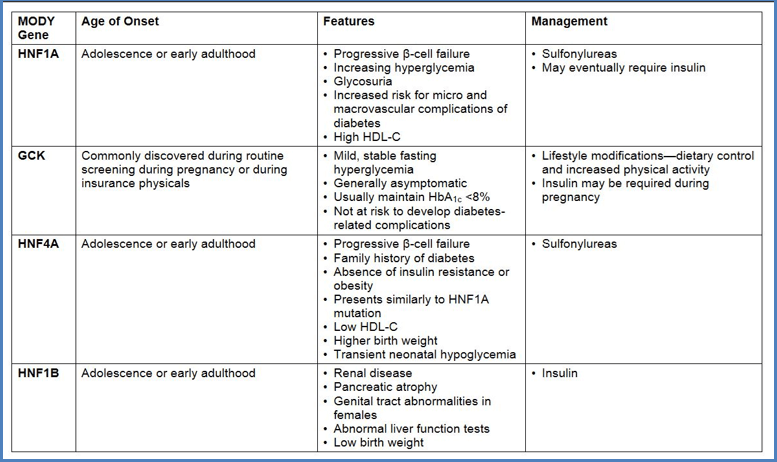
HNF1A-MODY (MODY 3):
- Definition: HNF1A-MODY is the second most common form of Maturity-Onset Diabetes of the Young (MODY).
- Inheritance Pattern: It follows an autosomal dominant inheritance pattern.
- Genetic Basis: HNF1A-MODY results from heterozygous pathogenic variants in the hepatocyte nuclear factor 1α (HNF1A) gene.
- Clinical Presentation: Patients with HNF1A-MODY often develop insulin-deficient diabetes during their second to third decade of life.
- Microvascular Complications: Without appropriate treatment, microvascular complications related to hyperglycemia are common.
- Treatment Approach: Sulfonylureas (SU): HNF1A-MODY patients typically respond well to SU medications.
- Early Recognition: Identifying HNF1A-MODY early in the disease course allows for targeted therapy.
- Cascade Testing: Recognition of one family member with HNF1A-MODY can prompt cascade testing in other family members, leading to significant therapeutic impact.
- Understanding the genetic basis of HNF1A-MODY informs personalized management decisions and improves outcomes.
HNF1B-MODY
(It is also known as MODY5 or renal cysts and diabetes (RCAD) syndrome)
- It is a complex disorder affecting multiple systems, with renal issues being more prevalent than diabetes. Renal involvement can manifest as structural defects detected either at birth or later in life. The most common structural abnormality is renal cysts, which may be observable prenatally as isolated bilateral hyperechogenic kidneys or postnatally as normal-size or small kidneys with hyperechogenicity and/or cysts. Other structural anomalies may include the absence of a kidney or renal hypoplasia.
- Functional renal defects can involve magnesium wasting, potentially leading to dangerous hypomagnesemia, and hyperuricemia, which may result in early-onset gout.
- The onset of diabetes mellitus is typically the primary extrarenal manifestation and often arises after the identification of renal disease in childhood. While the mean age of diabetes onset is around 24 years, it can occur anywhere from the neonatal period to late middle age. Other associated findings may include pancreatic atrophy, genital tract abnormalities in females, abnormal liver function, and primary hyperparathyroidism.
Clinical Pointers to MODY
| Consideration of MODY diagnosis: Onset of nonobese diabetes at a young age. Absence of signs of insulin resistance. Family history of diabetes spanning at least four generations. |
| MODY characteristics: Typically diagnosed in younger patients (<25 years). Absence of autoantibodies. Autosomal dominant transmission pattern. |
| MODY results from mutations in at least 14 different genes, leading to various pancreatic β-cell dysfunctions. |
| These mutations can affect pancreatic endocrine and exocrine functions, insulin gene structure, insulin secretion, and ATP-sensitive potassium channel function. |
| Among MODY cases: Mutations in the glycolytic enzyme glucokinase (GCK) account for approximately 32%. Hepatocyte nuclear factor genes (HNF1A, HNF4A, and HNF1B) mutations collectively represent around 68% of MODY cases, with HNF1A at 52%, HNF4A at 10%, and HNF1B at 6%. |
What is oligogenic form of diabetes?
- Initially, the field of genetics and diabetes primarily focused on highly penetrant, inherited forms of diabetes resulting from mutations in a single gene (monogenic diabetes).
- However, with the explosion of genomic data in recent years, attention has shifted toward conditions where two (digenic) or a few (oligogenic) genes with variant alleles contribute to an inherited form of diabetes. Often, these genes cluster in shared pathways, amplifying the impact of multiple genes that individually might not cause significant disease.
- For instance:
- A case of digenic MODY involves variants in both HNF1A and HNF1B genes.
- Severe insulin resistance can result from heterozygous variants in the PPARG and PPP1R3A genes.
- This broader perspective enhances our understanding of diabetes etiology and highlights the intricate interplay of genetic factors.
Syndromic diabetes:
- Patients with syndromic forms of diabetes exhibit a range of atypical clinical features alongside hyperglycemia.
- These unique combinations of syndromic features often result from a single molecular defect. Evaluation by a medical genetics expert is crucial.
- Genetic Etiology:
- Positive Family History: Syndromic diabetes may have a genetic basis, although severe cases can arise from de novo mutational events (appearing sporadic rather than familial).
- Gene Involvement: Genes associated with syndromic diabetes often encode proteins crucial for β-cell development, function, or survival.
- Multiorgan Involvement: Expression of these genes in other tissues leads to multiorgan effects.
- Examples of Syndromic Diabetes:
| Neurological Involvement: Microcephaly with simplified gyral pattern, epilepsy, and permanent neonatal diabetes syndrome (IER31P1 variants). |
| Other Endocrine Features: Neonatal diabetes mellitus with congenital hypothyroidism syndrome (GLIS3 variants). |
| Skeletal Involvement: Wolcott-Rallison syndrome (EIF2AK3 variants). |
| Gastrointestinal Involvement: Mitchell-Riley syndrome (RFX6 variants). |
| Immunological Involvement: Immunodysregulation, polyendocrinopathy, enteropathy X-linked syndrome (FOXP3 variants). |
LADA
- Latent Autoimmune Diabetes in Adults (LADA) usually presents in adulthood and progresses more slowly towards an absolute insulin requirement compared to type 1 diabetes mellitus. Despite the presence of pancreatic islet autoantibodies, the progression to beta-cell insufficiency in LADA is gradual.
- LADA is often confused with and misdiagnosed as ketosis-prone diabetes mellitus (KPD). Consequently, patients may be treated with oral hypoglycemic drugs, which can cause a more rapid decline in beta-cell reserve compared to KPD.
- Early initiation of insulin therapy is essential in LADA to delay rapid islet cell failure. The diagnosis of LADA is primarily based on the presence of glutamic acid decarboxylase (GAD) antibodies.
- Immunology of Diabetes Society has established three main criteria including:
- Adult age of onset (>30 years).
- Presence of any islet cell autoantibody.
- Absence of insulin requirement for at least 6 months after diagnosis.
- LADA is characterized by genetic, phenotypic, and immunological heterogeneity, with significant variability in the rate of β-cell destruction and different degrees of insulin resistance and autoimmunity. These differences are likely due to variations in genetic and immune factors.
Rule out secondary diabetes
- Pancreatic Disease:
- Cystic fibrosis
- Pancreatitis
- Alcoholism
- Hemochromatosis
- Pancreatectomy
- Endocrinopathies:
- Cushing’s syndrome
- Pheochromocytoma
- Glucagonoma
- Aldosteronoma
- Acromegaly
- Autoimmune diseases:
- Grave’s Disease
- Hashimoto’s Disease
- Addison’s Disease
- Chemical Diabetes:
- Glucocorticoids
- Thiazide diuretics
- Clonidine
- Lithium
- Oral contraceptives
- Phenothiazines
- Genetic Syndromes:
- Turner’s syndrome
- Leprechaunism
- Prader-Willi syndrome
Type 3c diabetes:
- Definition: Type 3c diabetes (also known as pancreatogenic diabetes) is diabetes that occurs secondary to pancreatic diseases, affecting both the exocrine and digestive functions of the pancreas. Researchers estimate that Type 3c diabetes represents 1% to 9% of all diabetes cases, but it is often misdiagnosed as Type 2 diabetes.
- Causes:
- Chronic Pancreatitis
- Cystic Fibrosis
- Pancreas Surgery (Pancreatectomy)
Ketosis-Prone diabetes:
- Ketosis-prone diabetes should be considered in individuals with phenotypic type 2 diabetes who present with insulin deficiency and ketoacidosis.
- Unlike classic type 1 diabetes, these patients may not have autoimmune markers (such as anti-GAD antibodies).
- Their condition can be transient, and some individuals regain beta-cell function after initial insulin therapy.
- It is now evident that there are at least four distinct phenotypes of ketosis-prone diabetes: A−B−, which is characterized by the absence of autoantibodies and β-cells; A+B−, denoting the presence of autoantibodies but absence of β-cells (typical of autoimmune Type 1 diabetes); A−B+, indicating the absence of autoantibodies but presence of β-cells; and A+B+, where both autoantibodies and β-cells are present.
- Case Scenario:
- A 19-year- male with no prior medical conditions presented to the emergency department after experiencing vomiting for one day. He reported symptoms of polyuria, polydipsia, and polyphagia for the past two weeks. With a family history of Type 2 diabetes in his maternal grandmother, his BMI was measured at 32 kg/m², and he displayed acanthosis nigricans on the back of his neck during physical examination. Initial laboratory tests revealed elevated plasma glucose levels at 1,100 mg/dL, decreased bicarbonate levels, an elevated anion gap, and a low arterial pH, consistent with diabetic ketoacidosis (DKA). His A1c was determined to be 13.2%, confirming new-onset diabetes.
- Following treatment for DKA, which included insulin glargine, rapid-acting insulin aspart, and metformin, the patient showed improvement upon follow-up one month later, displaying blood glucose levels within target ranges and occasional hypoglycemic episodes. Subsequent A1c readings indicated significant improvement, dropping to 5.0% at the one-month follow-up and further declining to 4.6% at the three-month mark. As a result, insulin doses were gradually reduced and eventually discontinued, with the patient maintaining normal blood glucose levels on metformin and lifestyle adjustments.
- Further assessments at the one-year follow-up revealed continued maintenance of normal A1c levels without the need for insulin, indicating successful management of the patient’s diabetes through metformin and lifestyle modifications. Additionally, tests for GAD-65 antibodies and islet-cell autoantibodies returned negative, further confirming the absence of autoimmune Type 1 diabetes.
Cystic fibrosis-related diabetes (CFRD)
- It is considered a spectrum of glucose tolerance abnormalities, starting with early-phase insulin secretion irregularities and progressing to declining β-cell function over time. The development of CFRD involves multiple factors. Timely identification and management are essential due to their significant impact on health outcomes. Currently, insulin therapy is the primary approach, but ongoing research aims to unravel the disease’s mechanisms for suggesting less intrusive treatment options in the future.
Bronze diabetes

A 51-year-old male presented with a diagnosis of type 2 diabetes (T2D).
- Family history: Negative for diabetes.
- Current treatments:
- Hypertension medication
- Metformin (1.5g/day)
- BMI: 31 kg/m²
- Waist circumference: 112 cm (consistent since his early 20s)
- HbA1c: 7.8%
The patient’s skin appeared reddish in color. Blood analysis revealed the following:
- Erythrocytosis:
- Hemoglobin (Hgb): 18.6 mg/mL
- Hematocrit (Hct): 53%
- Increased plasma ferritin: Up to 945 ng/mL
- Saturated transferrin fraction: >54%
- Transferrin: 255 µg/mL
- Consistent with a provisional diagnosis of hemochromatosis.
- Genetic analysis was carried out and showed the homozygous mutation H63D in the HFE gene.
Knowing lipodystrophic forms of diabetes:

- Clinical Characteristics:
- Loss of Body Fat: Lipodystrophic forms of diabetes are characterized by the loss of body fat.
- Generalized or Partial: This loss may be generalized or involve limited areas of the body (referred to as “partial lipodystrophy”).
- Accompanying Features:
- Prominent Musculature
- Phlebomegaly
- Severe Hypertriglyceridemia
- Profound Hepatomegaly (due to steatohepatitis)
- Hyperglycemia Driven by Insulin Resistance
- Severe Acanthosis Nigricans
Mitochondrial diabetes:
- Subtypes and Mitochondrial Dysfunction:
- Mitochondrial diabetes arises from mitochondrial dysfunction.
- Affected Organ Systems: Conditions often impact organs with high aerobic metabolism, such as the pancreas (especially β-cells).
- Inheritance Patterns:
- Maternal Inheritance: Variants encoded in the mitochondrial genome follow a strictly maternal inheritance pattern.
- Autosomal Recessive: Variants encoded in the nuclear genome exhibit autosomal recessive inheritance.
- Clinical and Prognostic Heterogeneity:
- Etiologic Variant Influence: Clinical variability results from the specific etiologic variant.
- Heteroplasmy or Homoplasmy: Degree of variant presence in mitochondrial genomes matters.
- Tissue Heterogeneity: Different tissues exhibit varying manifestations.
- Variant Example: c.3243A>G MT-TL1:
- Individuals with this variant may develop:
- Maternally Inherited Diabetes-Deafness Syndrome (MIDD)
- Mitochondrial Myopathy
- Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS)
- Leigh Syndrome
- Diabetes Response: May respond well to sulfonylureas or be characterized by insulinopenia.
- Age of Onset: Ranges from early childhood to ≥40 years, with some unaffected individuals.
- Individuals with this variant may develop:
Wolfram syndrome
- It is also referred to as diabetes insipidus, diabetes mellitus, optic atrophy, deafness syndrome (DIDMOAD), is typically caused by two faulty copies of the WFS1 or CISD2 genes, following an autosomal recessive inheritance pattern within families.
- The WFS1 gene produces wolframin, a transmembrane protein found in the endoplasmic reticulum. When wolframin functions abnormally, it induces endoplasmic reticulum stress, disrupting the unfolded protein response necessary for proper insulin synthesis. Consequently, Wolfram syndrome is recognized as one of the hallmark disorders associated with endoplasmic reticulum stress.
- Classic symptoms of Wolfram syndrome often manifest before the age of 16 and include insulin-deficient diabetes that doesn’t respond to antibodies. Additionally, patients typically experience optic atrophy, alongside potential sensorineural hearing loss, diabetes insipidus, urinary tract irregularities, progressive neurodegeneration, and psychiatric complications. Those with Wolfram syndrome caused by CISD2 variants tend to experience more severe symptoms and may also suffer from gastrointestinal bleeding.
Take Home Points:
- As our understanding of the pathophysiology of diabetes evolves, we increasingly recognize that many patients may have a form of diabetes that does not neatly fit with a diagnosis of either type 1 or type 2 diabetes.
- These forms of “atypical diabetes” have led to major contributions to our collective understanding of the basic biology that drives insulin secretion, insulin resistance, and islet autoimmunity.
- In fact, the frequency of non–type 1, non–type 2 diabetes—now referred to as “atypical diabetes”—is likely underestimated, and the proportion varies considerably depending on the ethnic and other characteristics of the population studied.
- When to Suspect Atypical Diabetes?
- Thorough Family History:
- Atypical diabetes can manifest in various ways, emphasizing the importance of a comprehensive family history.
- Autosomal Dominant Transmission: Many forms of monogenic diabetes follow an autosomal dominant inheritance pattern.
- Clues from Family Members: A family history of similar features across multiple generations prompts further testing.
- Presentation and Progression:
- Syndromic Forms of Diabetes:
- These present with manifestations specific to affected organ systems.
- Mitochondrial Diabetes: Patients may present with deafness or lactic acidosis.
- Pancreatogenic, Fulminant, and Slowly Progressive Forms: The patient’s medical history guides diagnosis.
- KPD Subtypes: A−β+ and A+β+ subtypes exhibit a striking history of initial DKA followed by insulin-free remission.
- Syndromic Forms of Diabetes:
- Physical Examination:
- Insulin Resistance Syndromes:
- Look for features of insulin resistance, adipose tissue disorders, and syndromes.
- Lipodystrophy Diagnosis: Identifying patterns of adipose tissue loss is crucial.
- Lipodystrophic Forms: Elevated triglycerides and low HDL cholesterol levels.
- Insulin Resistance Syndromes: Severe acanthosis nigricans and hirsutism are characteristic findings.
- Insulin Resistance Syndromes:
- Clinical Tests That Help Identify and Classify Forms of Atypical Diabetes:
- Islet Autoimmunity:
- The four most common autoantibodies in autoimmune diabetes are against GAD (GAD65), islet antigen 2 (IA-2), insulin, and zinc transporter 8 (ZnT8).
- Autoantibodies against insulin may be unreliable for patients already treated with exogenous insulin.
- Autoantibody Positivity: High titers of multiple islet autoantibodies suggest autoimmune forms of diabetes (e.g., T1D, LADA, or an “A+” subtype of KPD).
- The significance of a low titer of a single diabetes autoantibody remains unclear.
- Serum Insulin or C-Peptide Levels:
- Measure these as markers of the patient’s endogenous insulin secretory capacity.
- C-Peptide: Reflects residual β-cell capacity and is not part of therapeutic insulin preparations.
- Different cutoffs for fasting C-peptide levels determine adequate β-cell functional reserve.
- Oral Glucose Tolerance Test (OGTT):
- Evaluate insulin secretion dynamically.
- Calculate indices of insulin secretory capacity (e.g., C-peptide index (CPI)).
- Mixed-Meal Tolerance Test (MMTT): Stronger β-cell response due to modulation by incretin hormones.
- IDAA1c Measurement: Combines HbA1c and insulin dose to predict stimulated C-peptide levels.
- Probability Calculators for Monogenic Diabetes:
- Online tools aid clinical decision-making based on demographic and clinical information.
- Exeter MODY Probability Calculator: Helps determine the likelihood of monogenic diabetes.
- Pancreatic Diabetes and Insulin-Resistant Forms:
- Consider elevated fecal elastase for pancreatic diabetes.
- Rule out Cushing disease in insulin-resistant forms.
- Ordering and Interpreting Genetic Testing:
- Molecular Genetic Testing:
- Purpose: Molecular genetic testing is a powerful tool to diagnose single-gene atypical diabetes disorders and identify at-risk relatives.
- Recent Advances: Clinical guidelines for considering genetic testing in atypical diabetes have evolved.
- ISPAD Framework: The International Society for Pediatric and Adolescent Diabetes (ISPAD) provides a framework for testing well-defined monogenic forms of atypical diabetes (e.g., MODY and neonatal diabetes).
- Objective Assessment: Risk calculators (e.g., Exeter MODY risk calculator) offer an objective approach to assessing risk and stratifying patients likely to benefit from molecular testing.
- Genetic Testing Approaches:
- Broad-Based Molecular Tests: Exome sequencing (ES) and array comparative genomic hybridization demonstrate increased diagnostic utility compared to targeted gene panels.
- Methodology Choice: Sensitivity and specificity guide test selection based on suspected diagnosis.
- Practical Considerations: Gene panels and focused testing remain accessible due to cost and insurance coverage limitations for ES testing.
- Equitable Access: Implicit bias remains a barrier and should be minimized.
- Complex Results:
- Pretest Counseling: Address possible results, risks, benefits, and limitations with patients.
- Variant Classification: Follow standardized interpretation processes to classify variants (from “benign” to “pathogenic”).
- Variants of Uncertain Significance: These challenge clinical interpretation and actionability.
- Reclassification: New information can lead to more definitive variant classification.
CME INDIA Learning Edge
- Health care professionals can identify rare and atypical forms of diabetes by being attentive to unusual characteristics in their patients. For instance, they may question why a lean individual presents with apparent type 2 diabetes or why an obese patient displays symptoms resembling type 1 diabetes.
- A strong family history of diabetes with an early onset should raise suspicions of monogenic diabetes. Additionally, signs such as deafness, heart conditions, muscle disorders, and elevated blood glucose levels may suggest mitochondrial diabetes.
- While routine testing for islet antibodies and C-peptide levels is not common in adult patients, these tests can significantly aid in treatment decisions.
- Moreover, health care professionals should be familiar with criteria for genetic testing in cases where monogenic diabetes is suspected. Although genetic testing incurs a cost, advancements have made it more accessible and accurate than before.
- To determine when to order genetic testing, health care professionals can utilize pre-screening questionnaires or algorithms. These tools help guide decisions by considering factors like age of diabetes onset and the patient’s body composition. Online resources, such as the maturity-onset diabetes of the young (MODY) and diabetes subtypes calculator from the University of Exeter, offer likelihood percentages based on inputted information. If the calculator suggests a high likelihood of MODY, genetic testing is recommended.
- While predictive calculators aren’t flawless, they serve as valuable aids.
- Watch for:
| Diabetes in relation with Insulin Resistance |
| Genetic Defects in insulin action |
| Disease of exocrine pancreas |
| Diabetes in Endocrinopathies |
| Immune-mediated pathogenesis of diabetes |
| Diabetes of unknown cause |
| Diabetes arising in patients with genetic diseases |
| LADA |
| Ketosis prone diabetes |
CME INDIA Tail Piece
- Exeter MODY probability calculator (https://www.diabetesgenes.org/exeter-diabetes-app/ModyCalculator)

References:
- Parikh, Hemang M., et al. “Data mining framework for discovering and clustering phenotypes of atypical diabetes.” The Journal of Clinical Endocrinology & Metabolism 108.4 (2023): 834-846.
- Stephen I. Stone, Ashok Balasubramanyam, Jennifer E. Posey; Atypical Diabetes: What Have We Learned and What Does the Future Hold?. Diabetes Care 19 April 2024; 47 (5): 770–781. https://doi.org/10.2337/dci23-0038
- Chung, Wendy K., et al. “Precision medicine in diabetes: a consensus report from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).” Diabetes care 43.7 (2020): 1617-1635.
- Stone, Stephen I., et al. “Monogenic and syndromic diabetes due to endoplasmic reticulum stress.” Journal of Diabetes and its Complications 35.1 (2021): 107618.
- Akinci, B., Sahinoz, M., Oral, E., Feingold, K. R., Anawalt, B., Boyce, A., … & Wilson, D. P. (2000). Endotext. MDText.com, Inc., 2000
- Tamaroff J, Kilberg M, Pinney SE, McCormack S. Overview of Atypical Diabetes. Endocrinol Metab Clin North Am. 2020 Dec;49(4):695-723. doi: 10.1016/j.ecl.2020.07.004. Epub 2020 Oct 14. PMID: 33153675; PMCID: PMC8221417.

Discover CME INDIA

- Explore CME INDIA Repository
- Examine CME INDIA Case Study
- Read History Today in Medicine
- Register for Future CMEs
This paper are very informative.