

CME INDIA Presentation by Dr. K. Mugundhan, MD., DM (Neuro)., FRCP (Glasg, Lond, Edin & Ire), FACP, FICP, Professor of Neurology and Head, Stanley Medical College, Chennai.
Based on a presentation at APICON 2024, New Delhi.

First see this case
- A 50-yrs-old Driver admitted with C/O progressive loss of vision in Rt eye for 15 days.
- H/O flaccid weakness in both lower limb for 5 days.
- H/O loss sensation in both lower limbs below hip.
- H/O overflow incontinence of bladder present
- No H/O Trauma, Fever.
- Past History- No H/O similar illness in the past.
- Family History- Insignificant.
- Personal History- Non- Smoker, non – alcoholic, No extra marital contact.
What examination showed?
- Pt is conscious, oriented, afebrile, No neurocutaneous markers.
- Vital signs – stable.
- Central nervous system.
- HMF – Normal.
- Rt eye – Light perception absent.
- RAPD present.
- Lt eye – 6/6.
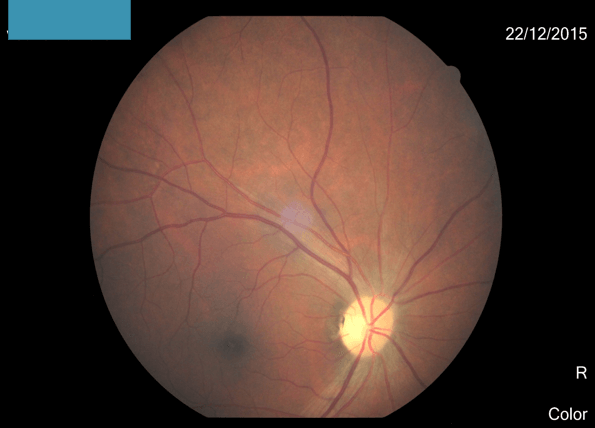
- Fundus- optic atrophy right side.
- Other cranial nerves are normal.
Spinomotor system
Upper Limb – normal
Trunk – weak
Lower Limb
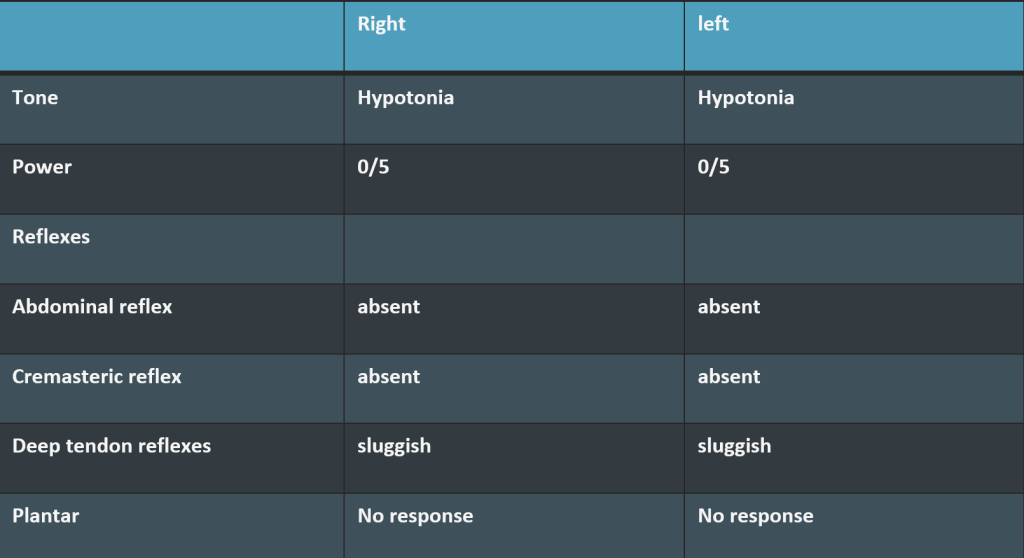
Sensory system: all modalities of sensations are lost below umbilicus.
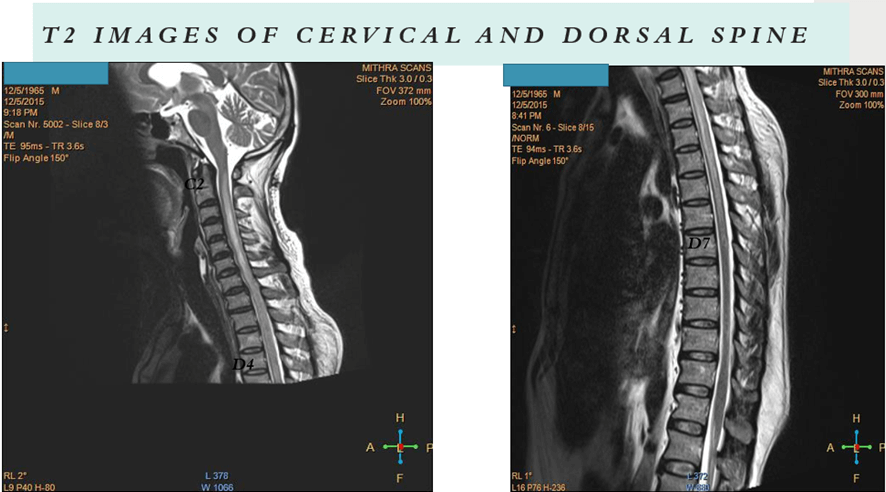
Demyelination (dorsal myelitis with right optic neuritis).
Investigations
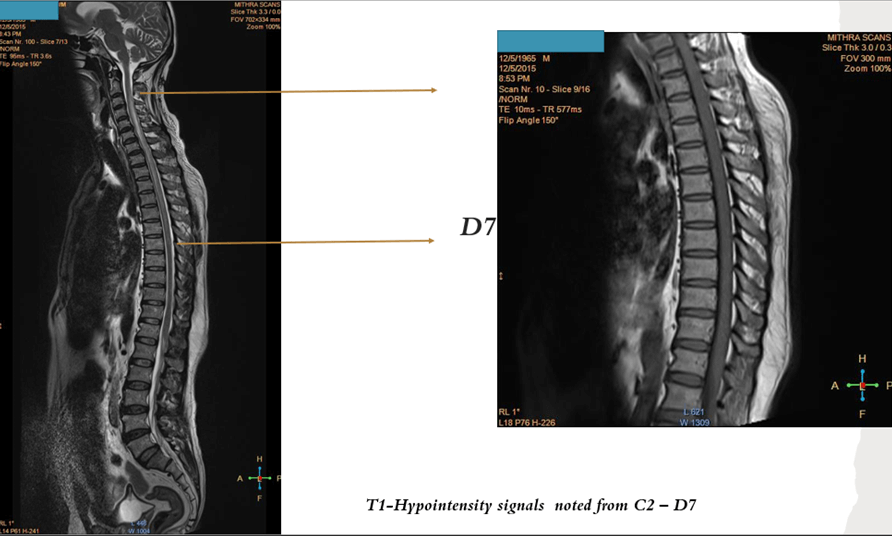
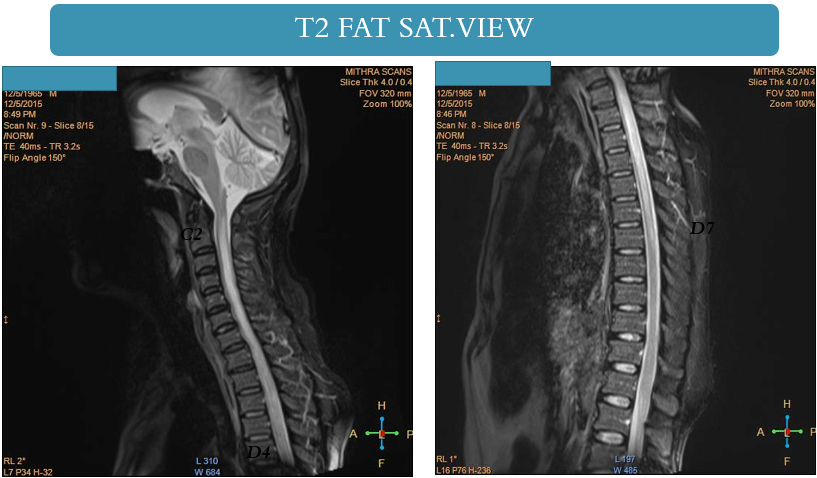
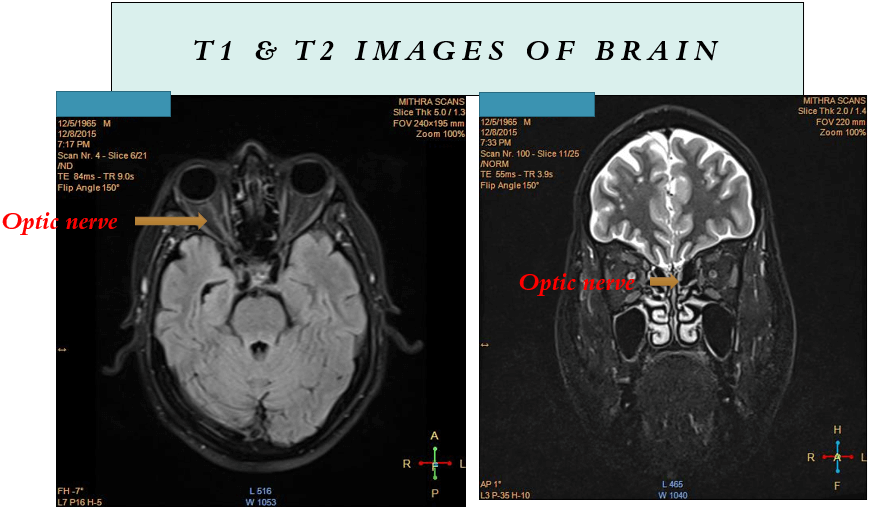
- MRI spine with brain including orbit
- Routine blood investigations including blood biochemistry were normal.
- Visual Evoked Potential (VEP)
- RT eye: could not be tested
- LT eye: normal
- MOG Igg antibody – Positive

Myelin oligodendrocyte glycoprotein (MOG)–associated disorders
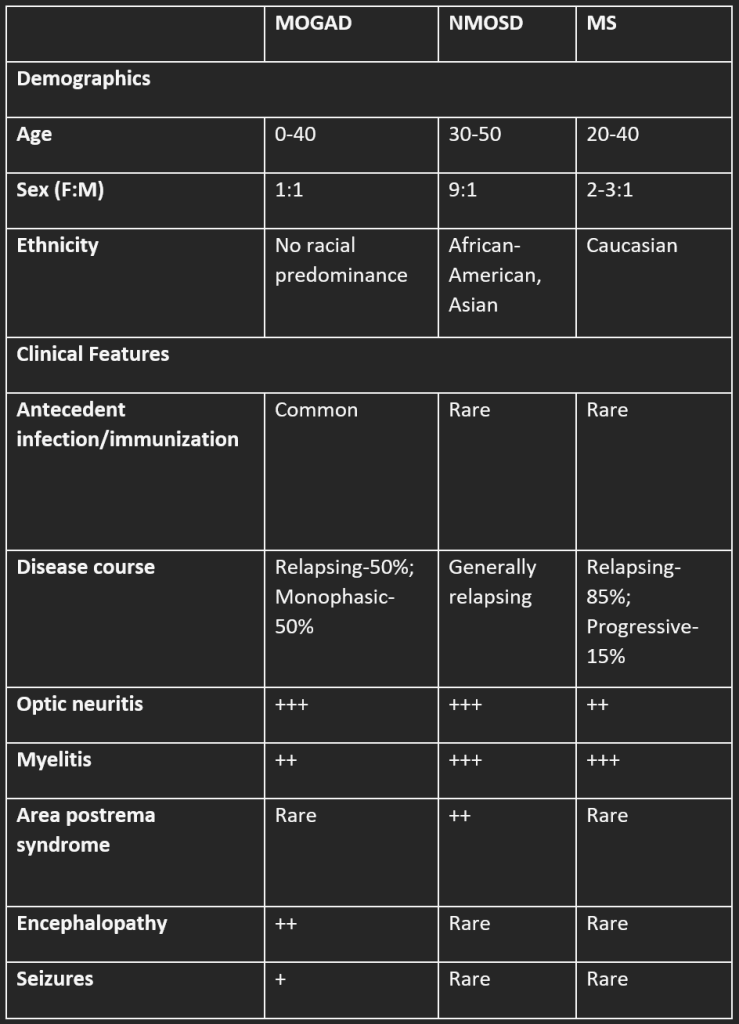
- These also referred to as MOGAD, emerged in the field of neuroimmunology relatively recently, gaining recognition as pathogenic entities around 2015. MOG represents a small proportion (0.05%) of central nervous system (CNS) myelin and is present on the outer lamella of the myelin sheath. Initially, MOG autoantibodies were believed to identify a subgroup of neuromyelitis optica (NMO) cases that lacked aquaporin-4 (AQP4). However, the understanding of MOG-associated disorders has evolved, and the clinical spectrum of these conditions is continuously broadening.
Epidemiology
- In contrast to multiple sclerosis (MS) and aquaporin-4 (AQP4)-positive neuromyelitis optica spectrum disorder (NMOSD), MOG-associated disorders do not exhibit a clear sex bias. Both males and females are affected similarly, with a higher incidence observed in children compared to adults.
- While the median age of onset typically occurs in the early to mid-thirties, the distribution of the disease shows two peaks, indicating a bimodal pattern. MOG-associated disorders can manifest as either a monophasic or relapsing course and may be triggered by viral infections or vaccination.
- Unlike AQP4-positive NMOSD and, to a lesser extent, MS, MOG-associated disorders are not strongly associated with other systemic autoimmune conditions, distinguishing them in this aspect.
Pathology
- The histological characteristics of MOG-associated disorders encompass perivenular and confluent white matter, as well as cortical demyelination. Notably, there is a heightened occurrence of intracortical demyelinating lesions accompanied by microglial infiltration. In contrast to the CD8+ lymphocyte dominance observed in multiple sclerosis (MS) lesions, MOG-associated disorders feature lymphocyte infiltrates primarily comprised of CD4+ cells.
- The pathobiology of MOG-associated disorders stands apart from AQP4-positive neuromyelitis optica spectrum disorder (NMOSD) by maintaining preserved astrocyte morphology and AQP4 expression, while lacking necrosis and extensive granulocytic cell infiltrates. This distinct set of histological features helps differentiate MOG-associated disorders from other neuroinflammatory conditions.
Clinical Phenotype
They include:
- Seronegative NMO (neuromyelitis optica)
- MS
- ADEM (acute disseminated encephalomyelitis)
- Transverse myelitis, and
- Chronic relapsing inflammatory optic neuritis.
The most common clinical manifestations are optic neuritis, ADEM, and myelitis, either alone or in combination.
Optic neuritis
- It stands out as the most prevalent manifestation, occurring in approximately 41% of pediatric patients and 56% of adult patients with MOG-associated disorders. This condition often presents as recurrent and bilateral, either simultaneously or sequentially with myelitis or another clinical phenotype associated with MOG.
- Clinical examination commonly reveals optic disc edema and peripapillary hemorrhages. Magnetic resonance imaging (MRI) frequently indicates longitudinally extensive optic nerve involvement with associated perineural enhancement, and there may be observable engagement of the optic chiasm in certain cases.
- Vision loss during the nadir is frequently severe for both MOG-associated disorders and aquaporin-4 (AQP4)-positive neuromyelitis optica spectrum disorder (NMOSD) optic neuritis. However, the recovery of visual acuity tends to be more favorable for MOG-associated disorders compared to AQP4-positive NMOSD. This distinction highlights the unique clinical characteristics of optic neuritis in MOGAD.
Myelitis
- It frequently emerges in conjunction with a prodromal illness in individuals with MOG-associated disorders. Magnetic resonance imaging (MRI) commonly reveals multiple spinal lesions, which can manifest as either short-segment or longitudinally extensive (involving three or more spinal segments). These lesions may affect the spinal cord anywhere from the medulla to the conus.
- Notably, gadolinium enhancement is infrequent in MOG-associated disorders. Disproportionate involvement of the spinal cord gray matter is a characteristic feature, presenting as an H pattern in the axial plane and a linear central T2 hyperintensity in the sagittal plane. Atrophy is rare, and imaging abnormalities in the spinal cord may show resolution following clinical recovery, underlining the dynamic nature of these manifestations in MOG-associated myelitis.
Neuromyelitis Optica Spectrum Disorder
- Between 7% and 42% of seronegative patients with NMOSD have anti-MOG auto antibodies and would be more accurately classified as having MOG-associated disorders.
- Simultaneous optic neuritis and myelitis or bilateral optic neuritis appear to be even more common for MOG-associated disorders than for AQP4-positiveNMO.
- Area postrema syndrome, however, is unusual for MOGAD.
Acute Demyelinating Encephalomyelitis
- Pediatric patients with MOG-associated disorders are highly likely to present with ADEM, especially those younger than 10 years of age.
- MRI typically reveals bilateral supratentorial brain lesions, which may affect both subcortical and deep white matter, along with involvement of the deep gray nuclei such as the basal ganglia and the thalamus.
- MOG autoantibody testing is warranted during the diagnostic workup for all pediatric patients with ADEM, particularly for those younger than 10 years of age.
Focal Encephalitis
- Rarely, MOGAD can present with focal encephalitis that may be associated with decreased consciousness, seizures/ status epilepticus, and focal weakness.
- MRI reveals hyperintense cortical FLAIR lesions that appear swollen and are usually unilateral.
- Corresponding leptomeningeal enhancement may be visible
- This clinical phenotype has been termed FLAMES (FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures).
Atypical Encephalitis
- A subset of encephalopathic patients with MOG autoantibodies do not meet ADEM criteria or exhibit a focal encephalitis syndrome.
- These patients present with altered consciousness, seizures, or a brainstem syndrome.
- MRI findings for patients with atypical encephalitis may include bilateral cortical MRI abnormalities and/or isolated deep gray matter involvement.
Chronic Lymphocytic Inflammation with Pontine Perivascular
Enhancement Responsive to Steroids (CLIPPERS)
- It is a rare syndrome that was first described in 2010.
- The diagnosis requires a clinical syndrome of sub-acute pontocerebellar dysfunction, without peripheral nerve involvement, that is responsive to steroids.
- On MRI, small, homogeneous gadolinium-enhancing nodules are visible in the pons and cerebellum with matching T2 lesions.
Diagnostic Work up
- A thorough diagnostic workup will include CNS imaging along with CSF and blood testing.
- Imaging in Myelin Oligodendrocyte Glycoprotein–Associated Disorders
- MRI remains one of the most clinically useful tools in evaluating acute autoimmune demyelinating syndromes, including MOG-associated disorders.
- Individuals with MOG-associated disorders exhibit a variety of distinctive radiologic characteristics that correspond to the clinical phenotype.
1. Acute optic neuritis-
- Perineural enhancement (50%)
- long segment lesions (80%)
- chiasmal involvement (10-20%)
2. Acute myelitis-
- Short-segment lesions and longitudinally extensive lesions both common
- multiple spine lesions
- central gray involvement (H sign) and linear sagittal T2 hyperintensity (~30%),
- involvement of the conus (4-40%);
- enhancement/ edema mild or relatively uncommon (25-70%)
Laboratory Findings in MOGAD
- MOG-associated disorders are defined by the presence of anti-MOG antibodies.
- These antibodies should be detected in the serum by using cell-based assays.
- Importantly, although MOG antibody titers greater than 1:20 are considered positive, the positive predictive value of testing increases with higher antibody titers.
- Titers of 1:20 to 1:40 have a positive predictive value of 51% for diagnosing MOG-associated disorders, whereas titers of 1:1000 or higher have a positive predictive value of 100%.
- CSF findings in MOG-associated disorders usually support the diagnosis of an inflammatory CNS syndrome.
- Pleocytosis is common during relapses and is often much more marked than that seen with MS or AQP4-positive NMOSD.
- 10% to 15% of patients with MOG-associated disorders have a CSF cell count of 100/mm3 or higher
- Although lymphocytes are the predominant cell type, neutrophils are also detectable in around half of specimens.
- CSF pleocytosis associated with MOG-associated disorders tends to be most marked during relapses, and CSF cell counts are often higher during ADEM or myelitis episodes as compared with isolated optic neuritis.
- CSF protein may be modestly elevated during relapses, although oligoclonal band is typically negative.

How to Manage?
- Acute attacks of MOG-associated disorders are treated similarly to acute events in other demyelinating syndromes.
- High-dose IV corticosteroids (20 mg/kg/d to 30 mg/kg/d methyl prednisolone for pediatric patients or 1000 mg/d methylprednisolone for adults) for 3 to 5 days are the first line of therapy, and most patients are steroid responsive.
- There appears an increased risk of relapses in the months following an acute episode. In particular, relapses may be seen on withdrawal of steroids.
- The use of a slow oral steroid taper for up to 3 months after an exacerbation can help to mitigate this risk.
- Pediatric guide lines suggest starting an oral prednisone taper at 1 mg/kg/d to 2 mg/kg/d (maximum 60 mg daily) and titrating down to 0.5 mg/kg/d over 1 to 4 weeks.
- Further prednisone tapering should occur more slowly, aiming to have the patient fully weaned by around 3 months after relapse. These guidelines may also be applied to adults.
- A subset of patients does not respond to steroids, and for these individuals either plasma exchange or IVIg can be used as second-line therapy during a relapse.
- Treatment escalation is warranted for patients who do not improve after intravenous methylprednisolone or individuals with a severe attack such as complete loss of vision, paralysis, or severe encephalopathy requiring admission to intensive care.
- Escalation therapies include plasma exchange (five exchanges on alternative days), immunoadsorption, intravenous immunoglobulins (total of 2 g/kg over 2 or 5 days), or plasma exchange followed by intravenous immunoglobulins.
- Controversy remains about when to start patients with MOG-associated disorders on long-term immunotherapy.
- As discussed, current natural history data support that a substantial subset of patients has monophasic disease, and committing these individuals to lifelong immunosuppression introduces unnecessary risks.
- A recent consensus statement from the European Union pediatric MOG consortium suggested initiating maintenance immunotherapy at the time of the second event (ie, first relapse).
- If subsequent relapses occur while on immunotherapy, an alternative agent should be considered.
- If relapses continue despite adequate treatment with at least two different immunomodulators, add-on therapy may be used;
- For example, oral prednisone or pulse IVIg could be added onto rituximab. These principles may also be applied for adults.
Prognosis
- 78% had “good” or full recovery from their initial episode.
- Younger age was positively associated with better recovery, whereas disability tended to be greater among patients who had experienced higher numbers of attacks and among those who did not fully recover from their onset attack.
CME INDIA Learning Points
- Over the past 15 years, there has been a significant shift in the global understanding of inflammatory demyelinating disorders within the central nervous system (CNS). This transformation arose with the recognition of distinct autoantibody-associated conditions, separate from multiple sclerosis (MS), namely aquaporin-4 (AQP4)-IgG-positive neuromyelitis optica spectrum disorder (AQP4-IgG+NMOSD) and myelin oligodendrocyte glycoprotein (MOG)-IgG-associated disease (MOGAD)
- Collaborative enhancements in the clinical-MRI characterization of these three disease entities have heightened diagnostic precision, enabling the identification of significant differences in pathophysiology, treatment response, and outcomes. Understanding the specific features that delineate each demyelinating disorder is essential for accurate diagnosis and the timely initiation of appropriate treatment
- MOG-associated disorders can manifest as either monophasic or relapsing, and the clinical course remains unpredictable at the onset. These disorders may emerge in a postinfectious context, but there is no strong association with a specific organism. MOG-associated disorders distinctly differ from both multiple sclerosis and aquaporin-4–positive neuromyelitis optica. Additionally, although rare, there may be occasional overlap with antibody-mediated autoimmune encephalitis.
- While rare phenotypic variants may still surface, the prevailing clinical presentations of MOG-associated disorders are now well-established and include optic neuritis, myelitis, and acute disseminated encephalomyelitis (ADEM). These manifestations can occur independently, sequentially, or in combination. Both children and adults can be affected by MOG-associated disorders, with ADEM being more prevalent in the youngest patients and myelitis in older individuals.
- In contrast to many other central nervous system demyelinating diseases, a subset of MOG-associated disorders follows a monophasic course. Ongoing research is essential to accurately identify patients prone to recurrent relapses and determine those who would benefit from maintenance immunotherapy.
- The treatment of MOG-associated disorder relapses may involve the administration of high-dose steroids, intravenous immunoglobulin (IVIg), and/or plasma exchange, with subsequent initiation of an oral steroid taper lasting up to 3 months. This extended taper is employed due to the temporal clustering of relapses.
- For relapsing MOG-associated disorders, steroid-sparing immunomodulators such as azathioprine, mycophenolate mofetil, rituximab, and IVIg have demonstrated efficacy. However, despite the use of these agents, many patients still experience some degree of breakthrough disease.
References:
- Sechi Elia , Cacciaguerra Laura , Chen John J. , Mariotto Sara , Fadda Giulia , Dinoto Alessandro , Lopez-Chiriboga A. Sebastian , Pittock Sean J. , Flanagan Eoin P. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management.Frontiers in Neurology,VOLUME=13,YEAR=2022 URL=https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2022.885218DOI=10.3389/fneur.2022.885218
- Sechi, Elia & Cacciaguerra, Laura & Chen, John & Mariotto, Sara & Fadda, Giulia & Dinoto, Alessandro & Lopez-Chiriboga, Alfonso & Pittock, Sean & Flanagan, Eoin. (2022). Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Frontiers in Neurology. 13. 10.3389/fneur.2022.885218.
- Hor JY, Asgari N, Nakashima I, et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. FrontNeurol 2020;11:501. doi:10.3389/fneur.2020.00501
- de Mol C, Wong Y, Pelt E, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. MultScler 2020;26(7):806-814. doi:10.1177/1352458519845112
- Ciotti JR, Eby NS, Wu GF, et al. Clinical and laboratory features distinguishing MOG antibody disease from multiple sclerosis and AQP4antibody-positive neuromyelitis optica. MultSclerRelatDisord 2020;45:102399. doi:10.1016/j.msard.2020.102399
- Dubey D, Pittock SJ, Krecke KN, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol 2019;76(3):301-309. doi:10.1001/jamaneurol.2018.4053
- Bruijstens AL, Wendel EM, Lechner C, et al. E.U.paediatric MOG consortium consensus: part 5 -treatment of paediatric myelin oligodendrocyteglycoprotein antibody-associated disorders. Eur J PaediatrNeurol 2020;29:41-53. doi:10.1016/j.ejpn.2020.10.005
- Longbrake E. Myelin Oligodendrocyte Glycoprotein-Associated Disorders. Continuum (Minneap Minn). 2022 Aug 1;28(4):1171-1193. doi: 10.1212/CON.0000000000001127. PMID: 35938661; PMCID: PMC9523511.

Discover CME INDIA

- Explore CME INDIA Repository
- Examine CME INDIA Case Study
- Read History Today in Medicine
- Register for Future CMEs